ARChE

Structure du noyau cellulaire avec les nucléoles (vert) et les centromères (rouge)
© Xavier Marques



Structure du noyau cellulaire avec les nucléoles (vert) et les centromères (rouge)
ADN Répété, Chromatine, Evolution
ADN Répété, Chromatine et Évolution (ARChE)
L’équipe ARChE se consacre à percer les mystères des séquences répétitives d’ADN présentes dans les génomes, en se penchant notamment sur leur impact dans l’organisation 3D du génome, dans la localisation des marques épigénétiques le long du génome, ainsi que dans l’expression génique dans différents modèles cellulaire. Une particularité de notre démarche scientifique vise aussi à étudier ces questions dans le contexte de l’évolution. Notre objectif est de mettre en lumière un potentiel rôle fonctionnel de ces séquences qui ont longtemps été considérées comme des vestiges de l’évolution ou comme des séquences d’ADN « égoïstes » et parasites.
Notre équipe interdisciplinaire se compose d’experts en bioinformatique évolutive et fonctionnelle (LP et JM), en chimie de l’ADN (CE), en microscopie à fluorescence (XM), en biologie moléculaire et en épigénétique (JL), ainsi qu’en culture cellulaire et en génie génomique (BDL). Elle compte en outre actuellement deux doctorants (MC et AW) et un ingénieur senior (TO) spécialisés en intelligence artificielle pour la génomique ainsi qu’un doctorant (JP) en biologie computationnelle évolutive. Nos recherches s’articulent autour de trois axes thématiques:
L’équipe étudie la diversité des séquences d’ADN alpha-satellite chez les singes du Vieux Monde, appelés cercopithécini, en élucidant sa distribution chromosomique qui fournit des informations sur l’histoire évolutive de ces séquences spécifiques aux primates. Nous avons utilisé des techniques de séquençage avancées et des méthodes d’analyse de données pour classifier des milliers de séquences, améliorant ainsi les modèles évolutifs des répétitions centromériques.
Développements futurs : Nous souhaitons étendre nos études à la compréhension de l’évolution des satellites alpha chez l’ensemble des primates. Nous souhaitons aussi adapter nos méthodes d’analyse pour commencer l’exploration de la diversité d’autres éléments répétitifs dans les génomes des primates, y compris les éléments transposables.

Analyse par FISH de la distribution de différentes familles de séquences alpha satellite sur des chromosome de Cercopithecus pogonias

Phylogénie simplifiée des primates
En utilisant des effecteurs de trans#Arche2cription similaires à des activateurs (TALEs) fusionnés à une déméthylase de lysine d’histone (KDM4B), l’équipe a réduit le niveau de H3K9me3 sur les répétitions alpha-satellites situées sur le chromosome 7 humain. Ces travaux ont apporté des éclairages sur la structure de la chromatine, le recrutement de protéines et la stabilité du centromère.
Développements futurs : Notre équipe a l’intention de contrôler la marque épigénétique H3K9me3 sur d’autres répétitions d’ADN et de mesurer ses effets sur l’organisation chromosomique, l’expression génique et la mémoire cellulaire. Nous utiliserons des outils optogénétiques pour obtenir un contrôle temporel précis des modifications épigénétiques.
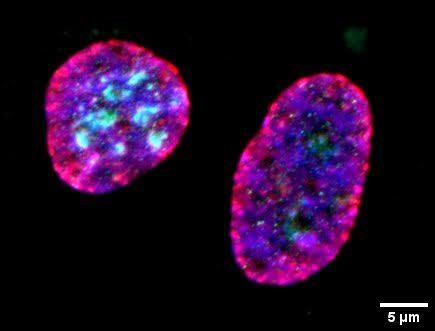
Marquage DAPI (bleu) Ki67 (vert) et hétérochromatine (H3K9me3, rouge)

Réécriture épigénétique des pénétromètres -> péricentromères
Notre équipe a exploité des techniques d’apprentissage profond pour annoter les génomes et prédire les marques fonctionnelles à partir des séquences d’ADN. Nous avons été les pionniers de l’approche « mutasome », qui consiste à muter chaque paire de bases d’un génome pour prédire l’effet sur l’annotation du génome. Ces travaux ont jeté les bases de l’écriture de génomes, c’est-à-dire la conception contrôlée de séquences aux propriétés spécifiques.
Développements futurs : Nous continuerons à exploiter les techniques d’apprentissage profond pour l’annotation et l’écriture de génomes, en nous concentrant sur la compréhension des séquences qui participent à la régulation des paysages épigénétiques. Nous nous attacherons aussi à étudier le positionnement des nucléosomes chez les mammifères, en mettant particulièrement l’accent sur les cellules souches embryonnaires de souris (collaboration avec P. Navarro) à l’institut Pasteur. Ces démarches visent aussi à mieux cerner le rôle des éléments répétitifs d’ADN dans le positionnement des nucléosomes et des marques épigénétiques qu’il portent dans le fonctionnement de notre génome, notamment dans la régulation des gènes dans différents types cellulaires..

Jouer au ping pong avec Enformer
L’approche interdisciplinaire de notre équipe intègre harmonieusement des méthodes expérimentales et computationnelles avancées, englobant le séquençage de pointe, la bioinformatique, la microscopie et l’édition du génome.
Inception
Bioconvergence
GP-write
Sorbonne centre for artifical inteligence (SCAI)
ANR
Institut Universitaire de France
GDR ADN
1: Routhier E, Joubert A, Westbrook A, Pierre E, Lancrey A, Cariou M, Boulé JB, Mozziconacci J. In silico design of DNA sequences for in vivo nucleosome positioning. Nucleic Acids Res. 2024 Jun 3:gkae468. doi: 10.1093/nar/gkae468. Epub ahead of print. PMID: 38828788.
2: Decombe S, Loll F, Caccianini L, Affannoukoué K, Izeddin I, Mozziconacci J, Escudé C, Lopes J. Epigenetic rewriting at centromeric DNA repeats leads to increased chromatin accessibility and chromosomal instability. Epigenetics Chromatin. 2021 Jul 28;14(1):35. doi: 10.1186/s13072-021-00410-x. PMID: 34321103; PMCID: PMC8317386.
3: Haschka T, Ponger L, Escudé C, Mozziconacci J. MNHN-Tree-Tools: a toolbox for tree inference using multi-scale clustering of a set of sequences. Bioinformatics. 2021 Nov 5;37(21):3947-3949. doi: 10.1093/bioinformatics/btab430. PMID: 34100911.
4: Routhier E, Pierre E, Khodabandelou G, Mozziconacci J. Genome-wide prediction of DNA mutation effect on nucleosome positions for yeast synthetic genomics. Genome Res. 2021 Feb;31(2):317-326. doi: 10.1101/gr.264416.120. Epub 2020 Dec 18. PMID: 33355297; PMCID: PMC7849406.
5: Cacheux L, Ponger L, Gerbault-Seureau M, Loll F, Gey D, Richard FA, Escudé C. The Targeted Sequencing of Alpha Satellite DNA in Cercopithecus pogonias Provides New Insight Into the Diversity and Dynamics of Centromeric Repeats in Old World Monkeys. Genome Biol Evol. 2018 Jul 1;10(7):1837-1851. doi: 10.1093/gbe/evy109. PMID: 29860303; PMCID: PMC6061836.


